ERYTHROBLASTOSIS FETALIS
 Erythroblastosis fetalis or hemolytic disease of the newton is a condition in which specific IgG antibodies formed by the mother are against erythrocyte antigens of the fetus; pass through the placenta by endocytosis mediated by receptors and are the fetal erythrocytes, which produces his abduction in the spleen, intravascular hemolysis, hemolytic anemia and unconjugated hyperbilirubinemia. This disease is caused by the progressive destruction of the erythrocytes of the fetus by antibodies against the Rh factor produced by a mother Rh-negative and that have been passed to the fetus through the placenta blood circulation. The predisposing factors include the transfusion or intramuscular injection of blood Rh-positive or that the intrauterine fetus is Rh-positive (which is the most common case).
Erythroblastosis fetalis or hemolytic disease of the newton is a condition in which specific IgG antibodies formed by the mother are against erythrocyte antigens of the fetus; pass through the placenta by endocytosis mediated by receptors and are the fetal erythrocytes, which produces his abduction in the spleen, intravascular hemolysis, hemolytic anemia and unconjugated hyperbilirubinemia. This disease is caused by the progressive destruction of the erythrocytes of the fetus by antibodies against the Rh factor produced by a mother Rh-negative and that have been passed to the fetus through the placenta blood circulation. The predisposing factors include the transfusion or intramuscular injection of blood Rh-positive or that the intrauterine fetus is Rh-positive (which is the most common case).
When the disease occurs with dropsy, the placenta produces a smooth and increased in size and ends with a dead newborn macerated. When the disease is of type jaundice occurs in newborns living with severe hemolytic anemia. In less severe cases; anemia is more mild, but the destruction of red blood cells also produce jaundice and increased bilirubin indirect.Some Rh negative women who conceived a child Rh-positive are treated with a protein marketed under the name of Rho GAM. The Rho GAM prevents the mother's body forming anti-Rh and, therefore, avoids the possibility of damage in case of a child following Rh-positive. Thus, the injection of Rh immunoglobulin to induce immune tolerance in the mother prevents erythroblastosis fetalis.
CAUSES OF THE ABO INCOMPATIBILITY
Type OThe people who have a blood type form proteins (antibodies) that cause the immune system to react against other blood types. The fact of being exposed to another type of blood can cause a reaction. This is important when a patient needs to receive an organ transplant or a blood transfusion. In these cases, the blood type must be compatible to avoid a reaction by the ABO incompatibility.


PERSONAL OPINION
Erythroblastosis fetalis, is a blood disorder in which a mother produces antibodies during pregnancy that attack the red blood cells in your own fetus this happens only when the mother has Rh-negative blood and the fetus Rh blood , inherited from the father. The reason for this problem is that the immune system of the mother holds the baby's Rh erythrocytes as strange, and are attacked by the maternal immune system.Treatment is based on intrauterine transfusions of red blood cells in the circulation of the baby after birth, treatment may include blood transfusions and intravenous fluids, respiratory support more closely through oxygen and phototherapy to destroy the excess bilirubin.Hypocalcemia in the newborn
NEONATAL HYPOCALCEMIA
DEFINITION
Defined as total serum calcium of less than 7 mg/dL (1.75 mmol/L) or ionized calcium less than 4 mg/dL(1 mmol/L) in preterm infants and less than 8 mg/dL (2 mmol/L; total) or <1.2 mmol/L (ionic) in term neonates [6]. The SCa concentration is usually reported in different ways viz. mg/dL, meq/L and mmol/L The relationship between these units is related to the following equations: mmol=L ¼ ½mg=dL_10__molecular wt;meq=L ¼ mmol=L_valency.Since the molecular weight of calcium is 40 and the valenceis +2, 1 mg/dL is equivalent to 0.25mmol/L and to 0.5meq/L. Thus, values in mg/dl may be converted to molar units (mmol/L) by dividing by 4.
Early onset neonatal hypocalcemia (ENH): This condition is fairly common and seen within the first 3-4 days of life in following clinical settings:Prematurity.This may be related to premature termination of trans-placental supply, exaggeration of the postnatal drop to hypocalcemic levels, increased calcitonin and diminished target organ responsiveness to parathyroid hormone.Infant of diabetic mother (gestational and insulin dependent).This may be related to increased calcium demands of a macrosomic baby. Magnesium depletion in mothers withdiabetes mellitus causes hypomagnesemic state in the fetus.This hypomagnesemia induces functional hypoparathyroidism and hypocalcemia in the infant. A high incidence of birth asphyxia and prematurity in infants of diabetic mothers are also contributing factors.Perinatal asphyxia Delayed introduction of feeds, increased calcitonin production, increased endogenous phosphate load, renal insufficiency, and diminished parathyroid hormone secretion all may contribute to hypocalcemia.
Maternal hyperparathyroidism. This causes intrauterine hypercalcemia suppressing the parathyroid activity in the fetus resulting in impaired parathyroid responsiveness to hypocalcaemia after birth. Hypocalcaemia may be severe and prolonged.Intrauterine growth restriction (IUGR) Infants with IUGR may have hypocalcemia if they are born preterm and/or have had perinatal asphyxia. Small for gestational age is not an independent risk factor for ENH.
Usually very nonspecific in the neonatal period, their potential serious complications, such as seizures, the laryngospasm or cardiac arrhythmias make this entity should be taken into account and searched in all newborns usually susceptible of autismDiagnosis1. Laboratory: Total or ionized serum calcium (total <7 mg/dL or ionized <4.0 mg/dL). Ionized calcium is the preferred mode for diagnosis of hypocalcemia.2. ECG: QoTc>0.22 s or QTc>0.45 sLate onset neonatal hypocalcemia (LNH)
This condition is rare as compared to ENH. It usuallypresents at the end of the first wk of life. It is usually symptomatic in the form of neonatal tetany or seizures.
Treatment of LNH

TREATMENT
1. Hypomagnesemia: Symptomatic hypocalcemia unresponsive to adequate doses of IV calcium therapy is usually due to hypomagnesemia. It may present either as ENH or later as LNH. The neonate should receive 2 doses of 0.2 mL/kg of 50% MgSO4 injection, 12 h apart, deep IM followed by a maintenance dose of 0.2 mL/kg/day of 50% MgSO4, PO for 3 days.
2. High phosphate load: These infants have hyperphosphatemia with near normal calcium levels. Exclusive breast-feeding should be encouraged and top feeding with cow’s milk should be discontinued. Phosphate binding gels should be avoided.
3. Hypoparathyroidism. These infants tend to be hyperphosphatemic and hypocalcemic with normal renal function. Elevated phosphate levels in the absence of exogenous phosphate load (cow’s milk) and presence of normal renal functions indicates parathormone inefficiency. It is important to realize that if the phosphate level is very high, then adding calcium will lead to calcium deposition and tissue damage. Thus, attempts should be made to reduce the phosphate (so as to keep the calcium and the phosphate product less than 55).These neonates need supplementation with calcium (50 mg/kg/day in 3 divided doses) and 1,25(OH)2 Vitamin D3 (0.5-1 μg/day). Syrups with 125 mg and 250 mg per 5 ml of calcium are available.1,25(OH)2 vitamin D3 (calcitriol) is available as 0.25 μg capsules. Therapy may be stopped in hypocalcemia secondary to maternal hyperparathyroidism after 6 wks.
4. Vitamin D deficiency states: These babies have hypocalcemia associated with hypophosphatemia due to an intact parathormone response on the kidneys. They benefit from Vitamin D3 supplementation in a dose of 30-60 ng/kg/day

PERSONAL OPINION
The hypocalcemia is the total serum calcium concentration lower than 8 mg/dl (ion 4 mg/dl) in the newborn to term and less than 7 mg/dl in the preterm infant. This is a metabolic disorder much more frequent in the neonatal period than at any other time in the life of the child, being a common cause of neonatal seizures their complications are serious, such as seizures, the laryngospasm or cardiac arrhythmias make this entity is to be taken into account and searched in all newborns usually susceptible to suffering mild hypocalcemia and/or the early onset of is often asymptomatic, while the late-onset usually appears with convulsions, the treatment that the baby can get (is) extra calcium, if necessary.
BIBLIOGRAPHY Ashish Jain & Ramesh Agarwal & M. JeevaSankar& Ashok Deorari& Vinod K. Paul. (27 July 2010 / Accepted: 2 August 2010 / Published online: 25 August 2010). Hypocalcemia in the Newborn. 08/10/15, de Indian J Pediatr Sitio web: http://medind.nic.in/icb/t10/i10/icbt10i10p1123.pdf
NEONATAL JAUNDICE
Jaundice refers to the yellow colouration of the skin and the sclerae (whites of the eyes) caused by the accumulation of bilirubin in the skin and mucous membranes. Jaundice is caused by a raised level of bilirubin in the body, a condition known as hyperbilirubinaemia.Approximately 60% of term and 80% of preterm babies develop jaundice in the first week of life, and about 10% of breastfed babies are still jaundiced at 1 month.For most babies, jaundice is not an indication of an underlying disease, and this early jaundice (termed 'physiological jaundice') is generally harmless.

Breastfed babies are more likely than bottle-fed babies to develop physiological jaundice within the first week of life.
Prolonged jaundice – that is, jaundice persisting beyond the first 14 days is also seen more commonly in these babies. Prolonged jaundice is generally harmless, but can be an indication of serious liver disease.Jaundice has many possible causes, including blood group incompatibility (most commonly Rhesus or ABO incompatibility), other causes of haemolysis (breaking down of red blood cells), sepsis (infection), liver disease, bruising and metabolic disorders.
Deficiency of a particular enzyme, glucose-6-phosphate-dehydrogenase, can cause severe neonatal jaundice. Glucose-6-phosphate-dehydrogenase deficiency is more common in certain ethnic groups and runs in families.Bilirubin is mainly produced from the breakdown of red blood cells. Red cell breakdown produces unconjugated (or 'indirect') bilirubin, which circulates mostly bound to albumin although some is 'free' and hence able to enter the brain. Unconjugated bilirubin is metabolised in the liver to produce conjugated (or 'direct') bilirubin which then passes into the gut and is largely excreted in stool. The terms direct and indirect refer to the way the laboratory tests measure the different forms. Some tests measure total bilirubin and do not distinguish between the two forms.In young babies, unconjugated bilirubin can penetrate the membrane that lies between the brain and the blood (the blood–brain barrier).
Unconjugated bilirubin is potentially toxic to neural tissue (brain and spinal cord). Entry of unconjugated bilirubin into the brain can cause both short-term and long-term neurological dysfunction (bilirubin encephalopathy). The term kernicterus is used to denote the clinical features of acute or chronic bilirubin encephalopathy, as well as the yellow staining in the brain associated with the former. The risk of kernicterus is increased in babies with extremely high bilirubin levels. Kernicterus is also known to occur at lower levels of bilirubin in term babies who have risk factors, and in preterm babies.Clinical recognition and assessment of jaundice can be difficult. This is particularly so in babies with darker skin tones.
Once jaundice is recognised, there is uncertainty about when to treat, and there is widespread variation in the use of phototherapy and exchange transfusion. There is a need for more uniform, evidence-based practice and for consensus-based practice where such evidence is lacking. This guideline provides guidance regarding the recognition, assessment and treatment of neonatal jaundice. The advice is based on evidence where this is available and on consensus-based practice where it is not.
TREATMENT
Treatment and care should take into account parents' preferences. Parents of babies with neonatal jaundice should have the opportunity to make informed decisions about their babies' care and treatment, in partnership with their healthcare professionals. If parents do not have the capacity to make decisions, healthcare professionals should follow the Department of Health's advice on consent and the code of practice that accompanies the Mental Capacity Act.
Pre-treatment
The enrolled infant will then have a TSB drawn using the Advanced Bilirubinometer Stat-Analyzer, Model BR2 (Advanced Instruments, Inc, Norwood, MA, USA) before treatment . Enrolled infants with elevated TSB qualify for inclusion into the second stage of thestudy and will be randomly assigned to receive FS-PT or CPT. Enrolled infants with TSB levels that are not elevated as defined above will not proceed to the second stage to be treated with PT. However, they will continue to be screened for elevated TcB as described above. If their subsequent TcB levels are not elevated during the first 14 days of life, the infant will be withdrawn from the study. All infants who meet the AAP guideline for the initiation of PT will be treated with CPT at night or when it is not possible to use FS-PT, secondary to rain or excessive cloud cover (defined as cloud cover persisting for more than 2 hours). FS-PT will be optimized in all infants by using white cloth lining the bottom and sides of the cot and exposing the infant maximally. CPT will be performed per international standards of practice.
Treatment
FS-PT will be started in the morning after the irradiance level inside the canopy is at least 8 μW/cm2 /nm, and will be stopped in the late afternoon when the irradiance level drops below 8 μW/cm2 /nm or at any point during the day during a rainy/cloudy day when the irradiance stays below 8 μW/cm2 /nm. If the irradiance persists below 8 μW/cm2 /nm for more than 1 hour and the infant qualifies for PT per the AAP guideline, CPT will be initiated. If the irradiance again exceeds 8 μW/cm2 /nm then the infant may again be placed in FS-PT.Treatment canopies Two FS-PT canopies will be used: one fitted with the Gila Titanium film and the other fitted with the Air Blue 80 film because of concerns about irradiance and heat. The Titanium canopy transmits approximately 33% blue light in the wavelength range 400 to 520 nm with much lower heat and will be used during sunny periods of the day. The Air Blue canopy transmits roughly 79% blue light as well as heat and will be used during cooler overcast periods of the day. The infants will be moved from one canopy to the other as needed during the day, when the weather changes, with the goal of keeping the irradiance level above 8 μW/cm2 /nm. The irradiance will be measured at an infant’s abdomen level with a BiliBlanket Meter II every half hour in infants under FS-PT (if possible, additional measurements may be done) and daily in those infants under CPT.
Treatment duration Generally, the infants will be placed under FS-PT/CPT from 9:00 am to 5:00 pm (target minimum duration of 5 hours per day for any infant in the study at 10:00 am, or 65% of total time between start time and 5:00 pm for infants enrolled after 10:00 am). The infants’ eyes will be protected with low-cost eye covers made from the elastic tops of any color socks. Bilirubin kinetics will only be calculated for infants who are able to spend at least 5 hours in FS-PT/CPT.

PERSONAL OPINION
Jaundice is a condition that causes a yellowing of the skin, in the tissues and fluids of the body, in newborn (neonatal) occurs when a baby has a high level of bilirubin in the blood. Bilirubin is a pigment that is released into the blood when destroying the red blood cells. The jaundice is normal in newborn infants because the babies are born with a greater number of red blood cells than they need, these red blood cells are destroyed and surplus release bilirubin in the blood, the goal of treatment is to prevent the elevation of bilirubin levels and the possible brain damage. However, it is important to remember that parents complications or damage caused by high levels of bilirubin are something very rare.Treatment decisions will depend on the age of the baby, whether or not you have other health problems and the speed with which they raise their levels of bilirubin.
BIBLIOGRAPHY
Tina M Slusher1,2*, Bolajoko O Olusanya3 , Hendrik J Vreman4 , Ronald J Wong4 , Ann M Brearley5 , Yvonne E Vaucher6 and David K Stevenson. (2013, 14:446). Treatment of neonatal jaundice with filtered sunlight in Nigerian neonates: study protocol of a non-inferiority, randomized controlled trial. 21/10/2015, de TRIALS Sitio web: http://www.trialsjournal.com/content/pdf/1745-6215-14-446.pdfClinical manifestation.
HYALINE MEMBRANE DISEASE
Respiratory
distress syndrome, also known as hyaline membrane disease, occurs almost exclusively
in premature infants
The
incidence and severity of respiratory distress syndrome are related inversely
to the gestational age of the newborn infant.
Pathogenesis:
respiratory distress syndrome develops because of impaired surfactant synthesis
and secretion leading to atelectasis, ventilation-perfusion (V/Q) inequality,
and hypoventilation with resultant hypoxemia and hypercarbia.
Blood gases
show respiratory and metabolic acidosis that cause pulmonary vasoconstriction,
resulting in impaired endothelial and epithelial integrity with leakage of
proteinaceous exudate and formation of hyaline membranes.
The
relative deficiency of surfactant decreases lung compliance and functional
residual capacity, with increased dead space.

Risk
factors:
- Prematurity
- Maternal diabetes
- Cesarean delivery
- Asphyxia
- White male infants
- Second-born twins
- Infants with a family history of respiratory distress syndrome
In contrast, the incidence of respiratory
distress syndrome decreases with the following:
- Use of antenatal steroids
- Pregnancy-induced or chronic maternal hypertension
- Prolonged rupture of membranes
- Maternal narcotic addiction
Clinical
presentation:
- Tachypnea
- Expiratory grunting
- Subcostal and intercostal retractions
- Cyanosis
- Nasal flaring
- Extremely immature in neonates may develop apnea and/or hypothermia
Differential Diagnoses:
- Anemia
- Aspiration pneumonia
- GERD
- Congenital pneumonia
- TTN
- Hypoglycemia
Fetal lung maturity tests: Prediction of fetal
lung maturity is derived by estimating the lecithin-to-sphingomyelin ratio
and/or by testing for the presence of phosphatidylglycerol in the amniotic
fluid obtained with amniocentesis.
Surfactant is a complex lipoprotein contains:
70-80% phospholipids, 8-10% protein, and 10% neutral lipids, primarily
cholesterol. Dipalmitoyl phosphatidylcholine (DPPC), or lecithin, is
functionally the principle phospholipid. Phosphatidylglycerol makes up 4-15% of
the phospholipids; although it is a marker for lung maturity, it is not
necessary for normal lung function.
Complications:
- Septicemia
- Bronchopulmonary dysplasia (BPD)
- Pulmonary hemorrhage
- Apnea/bradycardia
- Necrotizing enterocolitis
- ROP
- Failure to thrive
- Intraventricular hemorrhage (IVH)
Treatment
1. The use of antenatal steroids to enhance
pulmonary maturity
2. Appropriate resuscitation facilitated by
placental transfusion and immediate use of continuous positive airway pressure
(CPAP) for alveolar recruitment
3. Early administration of surfactant
4. The use of gentler modes of ventilation,
including early use of "bubble" nasal CPAP to minimize damage to the immature
lungs
5. Supportive therapies, such as the diagnosis
and management of PDA
6. Fluid and electrolyte management
7. Trophic feeding and nutrition

PERSONAL OPINION
The hyaline membrane disease is a disorder found in preterm infants, caused by insufficient production of surfactant together with the lack of development of the lungs. It can also be attributed to defective genes associated with the development of proteins linked to the production of pulmonary surfactant. The lungs show a deficiency in the production of surfactant, liquid allowing the alveoli to remain open during the pulmonary ventilation. The surfactant is a complex of lipids, proteins and glycoproteins produced by pneumocytes. Oxygen is administered with a small amount of CPAP plus intravenous fluids to stabilize the pressure and concentrations of sugar and blood salts. It has been shown that administration of artificial surfactant in newborns with hyaline membrane disease ventilated, a significant decrease of lethality is achieved by this disease.
Neonatal
Pneumonia
Pathogenesis
Pneumonia may be
acquired by intrauterine, intrapartum or postnatal routes.The pathogens include
mainly bacteria, followed by viruses and fungi which induce an inflammatory
pulmonary condition. This may cause epithelial injury to the airways, leakage
of proteinaceous fluid into the alveoli and interstitium, leading to surfactant
deficiency or dysfunction. Data from a German study suggest that respiratory insufficiency
in pneumonia is most likely caused by inhibition of surface-tension-lowering properties
of surfactant rather than by surfactant deficiency. Important predisposing
factors in the evolution of pneumonia are immaturity, low birth weight,
premature rupture of membranes, chorioamnionitis and factors associated with
prolonged neonatal intensive care.

Clinical
presentation, classification
Depending on the
time of manifestation of infection neonatal pneumonia may be classified as
early onset pneumonia (within the first 3 or 7 days of life, mostly within 48
hours), or late onset pneumonia (within 4 and 28 days of life). Congenital or
intrauterine pneumonia can be considered a variant of early onset pneumonia.
Other classifications refer to the underlying pathogen, like bacterial or viral
pneumonia or the pattern of lung infiltrates (e.g. interstitial pneumonia) on
chest radiographs. Clinical signs are unspecific and present as respiratory
distress of various degree, suspicious appearing tracheal aspirates, cough, apnea,
high or low temperature, poor feeding, abdominal distension, and lethargy.
Tachypnea is a
predominant clinical sign, present in 60-89 % of cases. Persistent fever is rather
unusual, but has been reported in neonates with viral pneumonia. The radiographical
appearance may also vary, showing reticulogranular-nodular infiltrates, and
bilateral streaky or hazy lungs. As small bronchioli tend to collapse there may
be compensatory hyperaeration in areas free of pneumonial infiltration. In
addition there may be pleural effusions and/or pneumatocele formation in more
complicated cases. Alveolar patterns with coarse, patchy parenchymal
infiltrates, consolidation, and diffuse granularity are more typical for
bacterial infections while parahilar streakiness, diffuse hazy lungs or reticulo-nodularity
are more common in viral disease. The differential diagnoses to be+ considered
on initial presentation are mainly surfactant deficiency syndrome and transient
tachypnoe of the newborn, in addition meconium aspiration syndrome (MAS),
pulmonary hemorrhage, pulmonary edema, primary pulmonary lymphangiectasis or
pulmonary lymphangiomatosis, congestive heart failure and Wilson-Mikity-syndrome.
Additional
investigations like echocardiography, high-resolution computed tomography, further
laboratory studies, and in rare cases lung biopsy are helpful in the diagnostic
work up.

Diagnosis
The clinical
diagnosis of pneumonia is challenging and may not always be correct (over- or underestimated).
Early tracheal aspirate cultures obtained within the first 8 to 12 hours of age
may help in diagnosing congenital pneumonia, especially in certain clinical
conditions, including maternal fever, clinical chorioamnionitis and leukopenia.
But even a positive blood culture or proven airway colonization do not
necessarily correlate with the clinical picture of sepsis or pneumonia. In the
clinical routine pneumonia is diagnosed based on a combination of perinatal
risk factors, signs of neonatal respiratory distress, positive laboratory studies,
radiological signs and a typical clinical course. Some clinical scenarios are more
or less suspicious. For example VAP, reported to be responsible for up to one
third of all nosocomial infections, may be suspected two or more days after the
initiation of mechanical ventilation when new or persistent infiltrates are
noticed in 2 or more chest radiographs. Additional definition criteria
developed by the Centers for disease control and prevention include an increase
in oxygen and ventilator requirements and at least three of the following signs
and symptoms: temperature instability, wheezing, tachypnea, cough, abnormal
heart rate, change in respiratory secretions, and abnormal peripheral white
blood count. The most common organisms in VAP in extremely preterm infants have
been shown to be Staphylococcus aureus and especially gram-negative organisms
like Pseudomonas aeruginosa , Enterobacter spp. and
Klebsiella spp.
Pneumonia caused by Ureaplasma species, Eubacteria mainly colonizing the
mucosal surface of the respiratory and urogenital tract, may be diagnosed by
direct isolation of the organism from endotracheal aspirates using culture or
PCR-techniques, by typical chest-x-ray patterns showing disseminated, patchy
infiltrates bilaterally with progression to cystic dysplasia, and elevated
inflammatory serum-parameters like CRP or an increased white cell count.
An organism
frequently associated with early onset pneumonia is Group B Streptococcus. The
clinical manifestation occurs usually within 6 of 8 hours of life and can
initially mimic surfactant deficiency syndrome.

Treatment,
prevention
As pneumonia is
often associated with or non distinguishable from bacterial sepsis initial therapy
at the NICU includes broad spectrum intravenous antibiotics according to local protocols.
In our unit we start with a combination of ampicillin and a second generation cephalosporine.
Although there is no evidence from randomized controlled trials that any antibiotic
regime is superior for suspected early onset neonatal sepsis, the WHO recommends
as first line treatment ampicillin plus gentamycin. In cases where we detect
pathogens in blood, or in endotracheal aspirates we treat according to susceptibility
from antiprogram results. A problem which is increasing worldwide in NICU´s is
the occurrence of multidrug resistant pathogens, mainly gram-negative bacilli.
As an alternative to systemic treatment aerosolized antibiotics like colistin
have been used successfully in patients with VAP caused by multidrug resistant
gram negative bacteria.
In patients
where we suspect or diagnose an U infection we initiate treatment with intravenous
clarithromycin (10mg/kg/day), a macrolid antibiotic. In a recently published randomized
controlled placebo single-center study clarithromycin treatment resulted in eradication
of Uu in 68,5 % of the patients and a significantly lower incidence of BPD
(2.9% vs. 36.4%) in preterm infants weighing between 750 to 1250 g .
Azithromycin, another macrolid antibiotic, which has good inhibitory activity against
Ureaplasma in in-vitro studies, may also be beneficial for BPD prevention in
Ureaplasma colonized/infected preterm infants, especially when used early and
for longer duration. In general the clinical and microbiological effectiveness
of macrolid antibiotics, the most commonly used in the literature being
erythromycin, has not yet been shown in adequately powered randomized controlled
clinical trials. Recommendations for the duration of antibiotic therapy in
proven neonatal pneumonia range from 10 to 21 days. Surfactant therapy may be
beneficial in selected patients by mechanisms improving lung function and
decreasing bacterial growth, but may require repeated doses. However, in a
recently published meta- analysis in patients > 35 weeks gestation with
proven or suspected pneumonia with onset during the first 28 days of life there
was no evidence of a significant effect on the primary outcome death, time to
resolution of pneumonia, BPD, pneumothorax and pulmonary hemorrhage. There are
still open questions related to the surfactant preparation, dosage, optimal
treatment frequency, number of doses and patient selection.
Severe cases of
pneumonia with respiratory insufficiency not responding to conventional therapy
may occasionally be candidates for ECMO. Preventative measures to be considered
include maternal infection control in the prenatal period, prenatal screening
and prophylaxis for streptococcal colonization, preference of non-or minimal
invasive procedures in the neonatal period like respiratory support without
intubation, immunoprophylaxis against RSV-infection, and general infection
control measures in the neonatal unit to reduce the incidence and transmission
of health-care-associated infections, the most important being hand hygiene
(38,39,40). Preventive strategies that may have a great impact are maternal and
infant vaccination programs, as has been already shown in developing countries
e.g for pneumococcal polysaccharide vaccines.
Early onset (< =7 days) Late onset (> 7 days)
Group B Streptococcus (g +) Escherichia coli (g-)
Escherichea coli (g-) Staphylococcus
epidermidis (g+)
Staphylococcus
aureus (g+) Klebsiella-Enterobacter-species
(g-)
Listeria
monocytogenes (g+) Pseudomonas
aeruginosa (g-)
Enterococcus (g +)
Ureaplasma urealyticum
(g+)*
g +/- =
gram-positive/negative
* based on
DNA-analysis

PERSONAL OPINION
Neonatal pneumonia is the most common serious infection in newborns, occurring mainly in preterm and men. Is a respiratory condition that has an index of neonatal deaths between 10 and 20%, neonatal pneumonia are inflammatory lesions are localized in the lung due to an infection, the cause of neonatal pneumonia are premature rupture of membranes over 12 h, vaginosis, intrapartum fever, foul amniotic fluid and pus, premature birth, treatment depends on the pathogen, early recognition of infection and early treatment before irreversible injury develops.
BIBLIOGRAPHY
Friedrich Reiterer. (2013). Neonatal Pneumonia. 06/11/2015, de doi Sitio web: http://dx.doi.org/10.5772/54310
Spina Bifida
Spina bifida is one of a group of birth defects known as neural tube defects (NTD). It occurs in the first month of fetal life, when the neural tube does not close properly, leaving an opening in the spinal cord and backbone. In California, spina bifida is found in about 1 in 2,860 pregnancies. There are three main types of spina bifida. The most serious form is called myelomeningocele, in which there is exposed tissue and nerves around the spinal cord. Meningocele occurs when the nerves and tissue form a cyst through the opening along the spine. The mildest form, called occulta, results from a small gap in one or more of the bones of the spine.
Children and young adults with spina bifida may have medical and developmental problems. Symptoms vary depending on the location of the defect. In severe forms of spina bifida hydrocephalus may also occur. Hydrocephalus is a condition in which there is extra fluid in the brain. Oftentimes, the inability to empty the bladder can lead to urinary tract disorders.

Causes, risk Factors
Mothers with the following characteristics have been found to be at higher risk for having a baby with spina bifida:
- Maternal lack of folic acid and vitamins
- Previous baby and family history of spina bifida
- Maternal obesity
- Diabetes during pregnancy
- Maternal stress
- Woman who has epileptic seizures
- Maternal fever in the first trimester
- Hispanic background

Prevention
Folic acid is a B vitamin that plays an important role in the development of the fetal brain and spinal cord during very early pregnancy, often before many women know they are pregnant. For this reason, and because many pregnancies are unplanned, it is recommended that all women of childbearing age take 400 micrograms of folic acid per day through dietary supplements or foods that are fortified with folate . It has been shown that the recommended dose reduces risk of spina bifida by 70%. Screening tests such as an ultrasound examination, a blood test called the maternal serum alphafetoprotein (MSAFP) or amniocentesis help identify fetuses at increased risk of an NTD . Women who think that they may be in a high risk category should have genetic counseling. For more information, please visit the California Prenatal Screening Program website.
There is no cure for spina bifida. However, surgery that occurs within the first days of life can assist and help improve the quality of life of these children. Generally, children with the mild form need no treatment. The key priority for treating spina bifida is to prevent infection (including meningitis) from developing through the exposed nerves and tissues . Thus, the fetus should be delivered at a facility that has personnel capable of handling all aspects of neonatal care . Complications of spina bifida range from minor physical problems to severe physical and mental disabilities. Other problems include paralysis or muscle weakness of the lower limbs, gastrointestinal disorders, and skin breakdown.

PERSONAL OPINION
The human nervous system develops from a small, specialized plate along the back of the embryo, cells early in development, this plate edges begin to curl and closer together, creating the neural tube, a narrow tube closes to form the brain and spinal cord of the embryo as development progresses, the top of the tube becomes the brain and the rest becomes the spinal cord. Spina bifida, which literally means "split spine" is characterized by incomplete development of the brain, spinal cord, or meninges (the protective covering around the brain and spinal cord).
Complications of spina bifida can range from minor physical problems to severe physical and mental disabilities.
BIBLIOGRAPHY
American College of Obstetricians and Gynecologists (ACOG). (2007). Screening for Birth Defects. Retrieved 10/11/2015, from www.acog.org/publications/patient_education/bp165.cfm
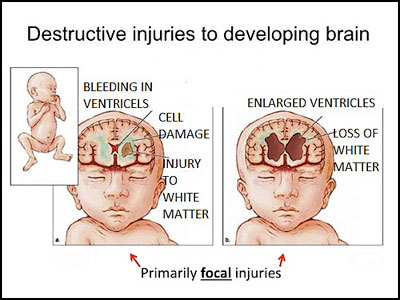
Periventricular Leukomalacia
Periventricular leukomalacia (PVL) is the
predominant form of brain injury and the leading known cause of cerebral palsy
and cognitive deficits in premature infants. The number of low-birth-weight
infants who survive to demonstrate these neurologic deficts is increasing.
Magnetic resonance imaging–based neuroimaging techniques provide greater
diagnostic sensitivity for PVL than does head ultrasonography and often
document the involvement of telencephalic gray matter and long tracts in
addition to periventricular white matter. The neuropathologic hallmarks of PVL
are microglial activation and focal and diffuse periventricular depletion of
premyelinating oligodendroglia. Premyelinating oligodendroglia are highly
vulnerable to death caused by glutamate, free radicals, and proinflammatory
cytokines. Studies in animal models of PVL suggest that pharmacologic
interventions that target these toxic molecules will be useful in diminishing
the severity of PVL.

Diagnosis of
Periventricular Leukomalacia
Periventricular leukomalacia is most often
diagnosed in the neonatal intensive care unit by means of head ultrasonography,
which demonstrates increased periventricular white matter echogenicity with or
without cystic abnormalities. Occasionally, PVL is detectable by means of
ultrasonography at birth or even in utero; however, cystic abnormalities often
do not become visible at ultrasonography until 1 week or longer after birth.
Periventricular leukomalacia can be all but excluded with normal findings at
postnatal cranial ultrasonographic examinations performed at 1 week and 1 month
after birth. In premature infants in whom repeated ultrasonography shows only
increased periventricular echogenicity without cysts, less than 5% will
subsequently develop overt cerebral palsy, although substantially more will
show evidence of cognitive dysfunction. The incidence of cerebral palsy, sometimes
complicated by refractory complex partial seizures, is much
Diagnosis of PVL is
generally discovered on a routine ultrasound to check for brain injury during
the hospital stay of a premature infant. There are few signs of PVL injury in
newborn and premature infants.
Early signs of PVL
that an infant may show are extreme stiffness or the inability or poor ability
to suckle. Severe PVL injury can be detected with the use of an ultrasound.
Magnetic resonance imaging (MRI) is more effective in the diagnosis of PVL.
Treatment and Prognosis of
Periventricular Leukomalacia
PVL treatment is only a response to the
individual needs of the patient. There is no treatment to cure PVL. Deficits
will need to be treated as they present and infants will need to be closely
monitored by a neurologist.
Cure PVL. Deficits will need to be treated
as they present and infants will need to be closely monitored by a neurologist.
Prognosis of periventricular leuklomalacia will depend on the extent of the damage
to the white matter of the brain. Some patients have minor deficits and
function well, while other patients have significant disabilities and deficits.
Types of deficits patients with PVL may
exhibit are developmental delays, deficits in posture, vision problems, motor
skill coordination difficulties, spastic diplegia, increased muscle tone,
spasticity in lower body, and gait while walking. Patients who suffer from
severe PVL may have high muscle tone and seizures and may be confined to a
wheelchair. Many infants suffering from severe PVL will develop cerebral palsy.
Patients diagnosed with PVL need to stay
informed and have frequent follow-up with their neurologists to discuss
treatment options and ongoing studies aimed at supporting and developing
improved treatment options for patients with PVL.

PERSONAL OPINION
PVL is a secondary pathological hypoxic injury to an ischemic episode damage creates holes in the brain. Usually appear premature condition involves the death of small areas of brain tissue around fluid-filled areas called ventricles, it is believed that a major cause of this condition are changes in blood flow to the area around the ventricles brain, there is no treatment for PVL. Cardiac activity kidney, lung, intestine and premature babies are carefully monitored.
BIBLIOGRAPHY
Progress
in Periventricular Leukomalacia, Wenbin Deng, MB, PhD; Jeanette Pleasure, MD; David Pleasure, MD March 2009 NEUROLOGICAL REVIEW Retrieved 20/11/2015. http://www.iprmd.org/downloads/publications/deng/14_Progress_in_Periventricular_Leukomalacia.pdf
CAH (Congenital Adrernal Hyperplasia)
CAH stands for
“congenital adrenal hyperplasia”. CAH is an inherited condition that
affects the adrenal glands and causes a
number of specific health issues.
‘Congenital’ means the
condition is present at birth. The adrenal glands are
cone-shaped organs that sit on top of each kidney. They make a number of
hormones necessary for healthy body function. Hyperplasia means ‘overly
large’. In people with CAH, the adrenal glands cannot make enough of a
hormone called cortisol. As they
start working harder in attempts to make more cortisol they increase in size,
resulting in hyperplasia.
Babies with CAH are
born with a number of physical changes. Their adrenal glands are often
larger than normal, even at birth. Girls with CAH may be born with external sex
organs that appear more masculine than they should. If not treated, both boys
and girls will develop early sexual characteristics, well before normal puberty
should begin.

Causes
Normally, the adrenal
glands make a number of different hormones, including cortisol, aldosterone and androgens.
Hormones are chemicals that send messages to other organs or tissues of the
body, telling them to do specific things.
CAH occurs when a
particular enzyme called
21-hydroxylase (21-OH) is missing or not working correctly. The job of this
enzyme is to help make cortisol and aldosterone in the adrenal glands so they can
be released when the body needs them.
One of the main jobs
of cortisol is to keep the amount of glucose, the sugar
used for energy by the body’s cells, at a normal level. Cortisol also
helps protect the body during times of physical or emotional stress such as
surgery, injury or illness. It helps to regulate the immune response and
inflammation so our bodies can deal with infection or illness.
Another hormone made
by the adrenal glands is aldosterone. This
hormone is released into the blood when the blood pressure drops too low.
It tells the kidneys to pull salt and water out of urine and put it back into
the blood. This raises blood pressure back to normal and prevents the body from
losing too much fluid. Babies with a form of CAH called “salt-wasting” do not
make enough aldosterone and they lose too much salt and water in their urine.
They become dehydrated and their blood pressure drops too low. This can
be life-threatening if not treated quickly.
The other hormones
made by the adrenal glands are called androgens. These
are male-like sex hormones. The adrenal glands also make a small amount
of female hormones.
Most people with CAH
make too much of the androgen hormones and not enough cortisol or
aldosterone. Having too much of the androgen hormones in the blood causes
female babies to develop masculine changes to their genitals. And, high
levels of androgens lead to early sexual development, well before the normal
age of puberty, in both boys and girls.
Problems
The effects of CAH can vary greatly from
person to person. There are a number of different forms of CAH which are
described below.
Most babies found to have CAH during newborn
screening have ‘classic CAH’. One type of classic CAH is called
‘salt-wasting’ which is a serious condition needing immediate treatment.
The other type of classic CAH is called ‘simple virilizing’. Children with this
type do not have immediate risks to their health but still need
treatment.
A small number of children are found through
newborn screening to have milder or ‘nonclassic CAH’ which often causes fewer
health problems. The symptoms of nonclassic CAH are quite variable from person
to person.
Classic CAH – “Salt-wasting form”
About 75% of babies with classic CAH have the ‘salt-wasting’ form. Salt-wasting CAH occurs when the adrenal glands make lower amounts of both cortisol and aldosterone and too much androgen. Babies who do not make enough aldosterone will start losing too much water and salt in their urine. This can quickly cause dehydration and very low blood pressure. This can be life-threatening if not treated right away.
About 75% of babies with classic CAH have the ‘salt-wasting’ form. Salt-wasting CAH occurs when the adrenal glands make lower amounts of both cortisol and aldosterone and too much androgen. Babies who do not make enough aldosterone will start losing too much water and salt in their urine. This can quickly cause dehydration and very low blood pressure. This can be life-threatening if not treated right away.
Infants with salt-wasting CAH usually show
some of the following features within the first few weeks of life:
·
Poor feeding
·
Listlessness and drowsiness
·
Vomiting
·
Diarrhea
·
Dehydration
·
Weight loss
·
Low blood pressure
·
Low blood salt (low blood sodium level)
If not treated, severe dehydration leads to shock, a
serious situation in which not enough blood is getting to the brain and other
organs. In babies with salt-wasting CAH, this is also called an "adrenal crisis”.
The signs of an adrenal crisis
include:
·
Confusion
·
Irritability
·
Rapid heart rate
·
Coma
Periods of adrenal
crisis due to too little aldosterone can occur as early as one week to one
month of age. If a child in shock is not treated, there is a risk of
death.
Even when carefully
treated, children with salt-wasting CAH are still at risk for adrenal crises
when they become ill or are under stress. The body needs more than the
usual amount of adrenal hormones during illness, injury or stress. This
means a child with CAH must be given more medication during these times to
prevent an adrenal crisis.
All babies with
salt-wasting CAH have the other features of classic CAH listed below.
Girls with salt-wasting CAH usually have more male-like changes to their genitals than girls
with simple virilizing CAH.
Classic CAH – Simple virilizing form
About 25% of babies with CAH have the simple virilizing form. The adrenal glands make enough aldosterone but not enough cortisol; they also make too much androgen.
Classic CAH – Simple virilizing form
About 25% of babies with CAH have the simple virilizing form. The adrenal glands make enough aldosterone but not enough cortisol; they also make too much androgen.
Classic CAH starts
its effects before birth. Excess androgen hormones are made by the fetus.
This causes the genitals of female
fetuses to develop male-like features. Baby girls born with classic CAH
often have an enlarged clitoris. In some
girls this is not very noticeable, but in others it may look like a small
penis. Baby girls may also have labia which are
fused together, may be wrinkled and may look more like a male scrotum. Some
baby girls have fewer genital changes than others. The high level of
androgen hormones does not affect the uterus and ovaries, which develop
normally.
Girls who are not
treated may develop other male-like traits and behaviors as they grow. Some of these changes may include:
- Deep, husky
voice
- Excess hair on the face and body
- Lack of menstrual periods or scanty or irregular
periods
- Early puberty changes such as hair in the armpits
and pubic area
- Severe acne
- Male-pattern baldness (loss of hair near the
temples)
Boys who are not
treated may have some of the following traits:
- Muscle growth at an early age
- Pubic hair and underarm hair during childhood
- Enlargement of the penis during childhood
- Early deepening of the voice
- Early beard
- Smaller than normal testicles
- Severe acne
Sometimes the changes
of early puberty happen in boys and girls as young as two to four years
old. Both boys and girls may have rapid growth during childhood but end
up being short as adults. Excess androgen hormones in childhood cause the rapid
growth. The androgens also cause shorter adult height by closing the growth plates too
soon.
Some untreated adults
also have problems with infertility and may have difficulty achieving
pregnancy.
Children with simple
virilizing CAH are at risk for adrenal crises, though typically less severe
than seen in children with the salt-wasting form. Acute illness or stress
increases the body’s need for cortisol. If children with CAH do not receive
increased amounts of medication during illness or stress, they are at risk for
health problems.
Nonclassic CAH /
Late-onset CAH
Nonclassic CAH, also called ‘late-onset’, usually causes milder effects than classic CAH. However, symptoms can be quite variable from person to person. Many people with nonclassic CAH often start showing signs during childhood, adolescence, or early adulthood. Some people never develop symptoms. Newborn screening can detect some, though not all, babies with the nonclassic form of CAH.
Nonclassic CAH, also called ‘late-onset’, usually causes milder effects than classic CAH. However, symptoms can be quite variable from person to person. Many people with nonclassic CAH often start showing signs during childhood, adolescence, or early adulthood. Some people never develop symptoms. Newborn screening can detect some, though not all, babies with the nonclassic form of CAH.
Babies with
nonclassic CAH are usually healthy at birth and their genitals are normal in
appearance. They do not have salt-wasting and are not at risk for adrenal
crises.
Children and adults
with nonclassic CAH have adrenal glands that make near-normal amounts of
cortisol and normal amounts of aldosterone. However, they have too much
17-OH progesterone (17-OHP), a chemical used to make cortisol, in their blood.
They also may make too much of the androgen hormones.
Some of the traits
that are sometimes seen in both males and females with nonclassic CAH include:
- Rapid growth in childhood and early teens with short adult height
- Severe acne
- Early puberty with development of pubic hair, underarm hair and body odor during childhood
- Excess hair on the face and other parts of the body
- Male-pattern baldness (hair loss near the temples)
Girls and women may
have:
- Male-like changes in physical appearance and behavior
- Irregular menstrual periods or early-onset of periods
- Infertility
Boys may have:
- Early beard growth
- Enlarged penis
- Small testicles

Treatment
Your baby’s primary doctor may work with a
pediatric endocrinologist to
provide medical care to your child. It is important for babies with
classic CAH to be diagnosed as quickly as possible. This allows treatment to
begin soon after birth which helps reduce the effects of CAH.
The main treatment for classic CAH is a drug
called ‘hydrocortisone’
(also called ‘cortisone’), taken
in pill form. This medication replaces the cortisol that your baby cannot
make on his or her own. It must be taken daily throughout life to prevent
effects of CAH. Cortisone is sometimes given in other drug form, such as
prednisone or dexamethasone
Treatment for Classic CAH – both simple
virilizing and salt-wasting forms:
Cortisone medication
The main treatment is to replace the amount of cortisol not being made by the adrenal glands. Hydrocortisone, a synthetic form of cortisol, is given by mouth in pill form. This treatment lessens the amount of androgens, prevents early puberty, and allows for more typical growth and development. Your doctor will follow your child’s growth, pubertal development, blood pressure, and hormone levels throughout childhood. The level of medication needed to control symptoms will be adjusted as needed throughout your child’s life.
The main treatment is to replace the amount of cortisol not being made by the adrenal glands. Hydrocortisone, a synthetic form of cortisol, is given by mouth in pill form. This treatment lessens the amount of androgens, prevents early puberty, and allows for more typical growth and development. Your doctor will follow your child’s growth, pubertal development, blood pressure, and hormone levels throughout childhood. The level of medication needed to control symptoms will be adjusted as needed throughout your child’s life.
It is important to always follow your
doctor's orders on how much cortisone to give your child. Too much
cortisone can cause temporary symptoms of Cushing syndrome so
the dose must be carefully balanced to your child’s height, weight and activity
level. Signs of Cushing syndrome include: stretch marks on the skin, rounded
face, weight gain, high blood pressure, and bone loss.
In addition, your doctor will give you
instructions for increasing the dose of hydrocortisone during an acute illness.
If you have questions about dosing, call your doctor. The body needs more
cortisol during illness, injury or times of stress. Therefore, the cortisone
dosage must be increased by your doctor when your child is ill, injured, or
requires surgery. If your child is ill and cannot take the pills, cortisone
injections may be necessary.
Your doctor may advise you to carry an
emergency treatment letter with steps for your child’s care during stress or
illness. Children with CAH should also consider wearing a Medic-Alert
bracelet.
Hydrocortisone must be taken throughout life
to prevent CAH effects. If the medication is stopped, symptoms will
develop.
Surgery for girls with classic CAH
Girls who are born with an enlarged clitoris or changes to the labia have the option of surgery to change their outer genitals to a more female appearance. Some women who have CAH have not had surgery and are happy they did not. Others are glad their parents decided to give them the surgery. This is a complex decision made by the parents with guidance from their doctors. Parents who are not sure about surgery may want to talk with other families who have faced similar decisions.
Girls who are born with an enlarged clitoris or changes to the labia have the option of surgery to change their outer genitals to a more female appearance. Some women who have CAH have not had surgery and are happy they did not. Others are glad their parents decided to give them the surgery. This is a complex decision made by the parents with guidance from their doctors. Parents who are not sure about surgery may want to talk with other families who have faced similar decisions.
If you choose corrective surgery, it can be
done as early as age one to three. Surgery on the clitoris usually hides
the excess tissue but leaves the clitoris itself intact. Surgery to
separate the labia and to create a normal vagina is often delayed until the
teenage years. Ask your doctor about the risks and benefits of surgery
for these changes and the best time to do these surgeries.
Treatment to prevent short stature
Your doctor may take periodic X-rays to check your child’s ‘bone age’. This allows your doctor to tell whether your child is growing at too rapid a rate. It also shows whether the growth plates are still open or whether they are closing too early.
Your doctor may take periodic X-rays to check your child’s ‘bone age’. This allows your doctor to tell whether your child is growing at too rapid a rate. It also shows whether the growth plates are still open or whether they are closing too early.
Specific medications may help increase height
in children and teens that show signs of early growth failure. Certain
medications lower androgen levels. If you have questions about your
child’s growth, talk to your doctor about the costs and benefits of these
treatments.
Treatment for early puberty
Children who show changes of puberty at a young age are sometimes treated with medications that lower the amount of androgen hormones. Your doctor will talk to you about these medications should your child start showing signs of puberty during childhood.
Children who show changes of puberty at a young age are sometimes treated with medications that lower the amount of androgen hormones. Your doctor will talk to you about these medications should your child start showing signs of puberty during childhood.
Additional treatment for classic CAH –
salt-wasting form
Children with salt-wasting CAH need to take an additional medication called Florinef. Florinef (9a-fludrohydrocortisone) is a ‘salt-retaining’ drug that replaces the aldosterone absent in children with salt-wasting CAH. It is given by mouth in pill form.
Children with salt-wasting CAH need to take an additional medication called Florinef. Florinef (9a-fludrohydrocortisone) is a ‘salt-retaining’ drug that replaces the aldosterone absent in children with salt-wasting CAH. It is given by mouth in pill form.
Some children with salt-wasting CAH need to
follow a food plan that contains more salt than usual. In addition, your
doctor may recommend salt tablets to prevent dehydration. It is important
to follow your doctor’s instructions on how much salt to feed your child.
Most children on medication do not need to add extra salt to their diets.
Treatment for nonclassic CAH
Some people with nonclassic CAH do not need treatment and may go through life without symptoms. Others begin having symptoms in childhood, adolescence or young adulthood and may need medication in the form of cortisone pills. Symptoms that may signal the need for treatment include:
Some people with nonclassic CAH do not need treatment and may go through life without symptoms. Others begin having symptoms in childhood, adolescence or young adulthood and may need medication in the form of cortisone pills. Symptoms that may signal the need for treatment include:
·
Severe acne
·
Excess body hair
·
Irregular menstrual periods
·
Lumps in the testicles
·
Infertility
Children and adults with nonclassic CAH
usually need less medication than children with classic CAH.

PERSONAL OPINION
Children who have two adrenal glands, one located on top of each kidney. These glands produce cortisol and aldosterone hormones that are essential for life. People with congenital adrenal hyperplasia lack an enzyme needed the adrenal gland to produce hormones, the symptoms vary depending on the type of CAH have someone of his age and when the disorder is diagnosed, the goal of treatment is to return hormone to normal or near normal levels.
BIBLIOGRAPHY
MercSeromo . (MET-AUG-11-ANZ-08). Congenital Adrenal. 27/15/2015, de Congenital Adrenal Hyperplasia Sitio web: http://www.apeg.org.au/Portals/0/resources/Hormones_and_Me_8_CAH.pdf
Impetigo
I mpetigo is a common bacterial skin infection
caused by Staphylococcus aureus, group A beta-hemolytic Streptococcus pyogenes,
a combination of the two, or less commonly, anaerobic bacteria.1,2 In the
United States, more than 11 million skin and soft tissue infections are caused
by S. aureus annually.3 Impetigo is the most common skin infection in children
two to five years of age, but persons of any age can be affected.4 One-third of
skin and soft tissue infections in returning travelers are attributable to
impetigo, usually secondary to infected mosquito bites.5 Many bacteria inhabit
healthy skin; some types, such as S. pyogenes and S. aureus, intermittently
colonize the nasal, axillary, pharyngeal, or perineal areas.2,6 These bacteria
can lead to infection of susceptible skin.

Complications.
Impetigo is usually a self-limited condition,
and although rare, complications can occur. These include cellulitis
(nonbullous form), septicemia, osteomyelitis, septic arthritis, lymphangitis,
lymphadenitis, guttate psoriasis, staphylococcal scalded skin syndrome, and
acute poststreptococcal glomerulonephritis, with poststreptococcal glomerulonephritis
being the most serious. Most cases of poststreptococcal glomerulonephritis in
the United States are associated with pharyngitis. The strains of S. pyogenes
implicated in impetigo are thought to have minimal nephritogenic potential.

Treatment
The aims of treatment include relieving the
discomfort and improving cosmetic appearance of the lesions, preventing further
spread of the infection within the patient and to others, and preventing
recurrence. Treatments ideally should be effective, inexpensive, and have
limited side effects. Topical antibiotics have the advantage of being applied
only where needed, which minimizes systemic side effects. However, some topical
antibiotics may cause skin sensitization in susceptible persons. A Cochrane
review of interventions for impetigo identified only 12 good-quality studies
of impetigo treatment.8 In a 2003 meta-analysis that included 16 studies, 12
received a good-quality score.4 Most of the studies addressed nonbullous
impetigo, although the limited data for bullous and common impetigo suggest
that similar conclusions may be drawn regarding treatment
Topical antibiotics versus placebo
Three studies found that topical antibiotics
are clearly more effective than placebo for the treatment of impetigo.4,8 Most
patients with localized disease should receive mupirocin (Bactroban) or fusidic
acid (not available in the United States) because they are effective and well
tolerated. Data from four trials show that they are equally effective.4,8 Data
on other topical antibiotics were limited, but bacitracin and
bacitracin/neomycin were less effective. Adverse effects from topical
antibiotics were uncommon and, when present, were mild.
Oral antibiotics
Oral penicillin V was no more effective than
placebo in a single study of patients with impetigo; however, the study was too
small (and therefore lacked adequate statistical power) to show a clinically
meaningful difference between the treatment and placebo groups, if one
existed.8 Data comparing other oral antibiotics with placebo are not available.

PERSONAL OPINION
Impetigo is a superficial skin infectious disease caused by bacteria, which occurs most often in children. It is classified into primary impetigo when it comes to direct bacterial invasion of previously normal skin, or secondary or common impetigo, when the infection is secondary to other underlying skin diseases that affect the skin barrier, such as scabies or eczema expectant management pending a native resolution only performing hygiene, topical disinfectants, such as saline, hexachlorophene, povidone-iodine and chlorhexidine.
Este comentario ha sido eliminado por el autor.
ResponderBorrarEste comentario ha sido eliminado por el autor.
ResponderBorrarEste comentario ha sido eliminado por el autor.
ResponderBorrarIrrespective of receiving daily oral or future injectable depot therapies, these require health care visits for medication and monitoring of safety and response. If patients are treated early enough, before a lot of immune system damage has occurred, life expectancy is close to normal, as long as they remain on successful treatment. However, when patients stop therapy, virus rebounds to high levels in most patients, sometimes associated with severe illness because i have gone through this and even an increased risk of death. The aim of “cure”is ongoing but i still do believe my government made millions of ARV drugs instead of finding a cure. for ongoing therapy and monitoring. ARV alone cannot cure HIV as among the cells that are infected are very long-living CD4 memory cells and possibly other cells that act as long-term reservoirs. HIV can hide in these cells without being detected by the body’s immune system. Therefore even when ART completely blocks subsequent rounds of infection of cells, reservoirs that have been infected before therapy initiation persist and from these reservoirs HIV rebounds if therapy is stopped. “Cure” could either mean an eradication cure, which means to completely rid the body of reservoir virus or a functional HIV cure, where HIV may remain in reservoir cells but rebound to high levels is prevented after therapy interruption.Dr Itua Herbal Medicine makes me believes there is a hope for people suffering from,Parkinson's disease,Schizophrenia,Lung Cancer,Breast Cancer,psoriasis,Colo-Rectal Cancer,Blood Cancer,Prostate Cancer,siva.Fatal Familial Insomnia Factor V Leiden Mutation ,Epilepsy Dupuytren's disease,Desmoplastic small-round-cell tumor Diabetes ,Coeliac disease,Creutzfeldt–Jakob disease,Cerebral Amyloid Angiopathy, Ataxia,Arthritis,Amyotrophic Lateral Scoliosis,Fibromyalgia,Fluoroquinolone Toxicity
ResponderBorrarSyndrome Fibrodysplasia Ossificans ProgresSclerosis,Alzheimer's disease,Adrenocortical carcinoma.Asthma,Allergic diseases.Hiv_ Aids,Herpe ,Copd,Glaucoma., Cataracts,Macular degeneration,Cardiovascular disease,Lung disease.Enlarged prostate,Osteoporosis.Alzheimer's disease,
Dementia.(measles, tetanus, whooping cough, tuberculosis, polio and diphtheria),Chronic Diarrhea,
Hpv,All Cancer Types,Diabetes,Hepatitis,I read about him online how he cure Tasha and Tara so i contacted him on drituaherbalcenter@gmail.com / info@drituaherbalcenter.com. even talked on whatsapps +2348149277967 believe me it was easy i drank his herbal medicine for two weeks and i was cured just like that isn't Dr Itua a wonder man? Yes he is! I thank him so much so i will advise if you are suffering from one of those diseases Pls do contact him he's a nice man.